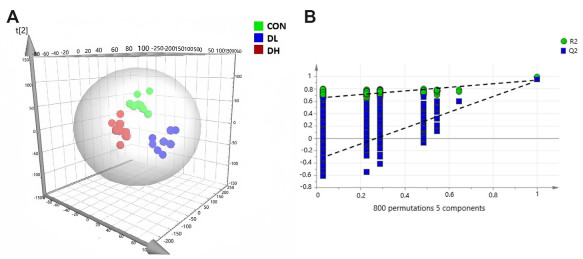
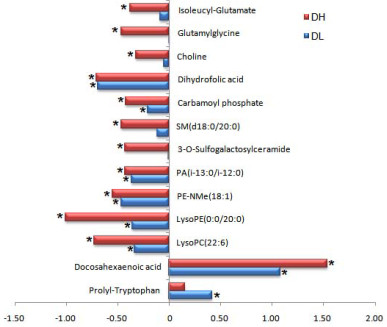
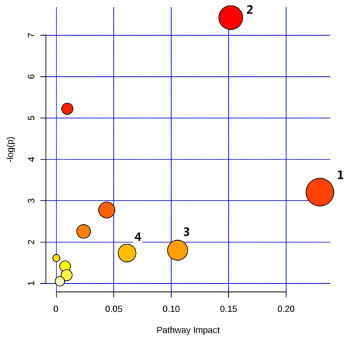
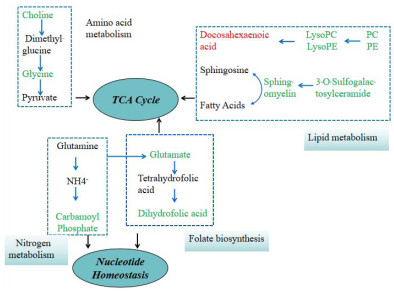
| Citation: | Zhen Kang, Qianqi Hong, Fei Yan, Tianyi Yu, Yuna Bai, Xiaobo Liu, Xiaolin Na, Cheng Wang. Metabolomics study on dibenz[a, h]anthracene exposure-induced pulmonary injury in rats after intratracheal instillation[J]. Frigid Zone Medicine, 2025, 5(1): 42-49. doi: 10.1515/fzm-2025-0004

|
| [1] |
Patel A B, Shaikh S, Jain K R, et al. Polycyclic aromatic hydrocarbons: sources, toxicity, and remediation approaches. Front Microbiol, 2020; 11, 562813. doi: 10.3389/fmicb.2020.562813
|
| [2] |
Hertz-Picciotto I, Baker R J, Yap P S, et al. Early childhood lower 48 respiratory illness and air pollution. Environ Health Perspect, 2007; 115(10): 1510-1518. doi: 10.1289/ehp.9617
|
| [3] |
Jeng H A, Pan C H, Diawara N, et al. Polycyclic aromatic hydrocarbon-induced oxidative stress and lipid peroxidation in relation to immunological alteration. Occup Environ Med, 2011; 68(9): 653-658. doi: 10.1136/oem.2010.055020
|
| [4] |
Perera F P, Li Z, Whyatt R, et al. Prenatal airborne polycyclic aromatic hydrocarbon exposure and child IQ at age 5 years. Pediatrics, 2009; 124(2): e195-e202. doi: 10.1542/peds.2008-3506
|
| [5] |
Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol, 2011; 31(3): 363-373. doi: 10.1016/j.reprotox.2010.12.055
|
| [6] |
Wassenberg D M, Di Giulio R T. Synergistic embryotoxicity of polycyclic aromatic hydrocarbon aryl hydrocarbon receptor agonists with cytochrome P4501A inhibitors in Fundulus heteroclitus. Environ Health Perspect, 2004; 112(17): 1658-1664. doi: 10.1289/ehp.7168
|
| [7] |
Kim A, Park M, Yoon T K, et al. Maternal exposure to benzo[b] fluoranthene disturbs reproductive performance in male offspring mice. Toxicol Lett, 2011; 203(1): 54-61. doi: 10.1016/j.toxlet.2011.03.003
|
| [8] |
Kummer V, Mašková J, Matiašovic J, et al. Morphological and functional disorders of the immature rat uterus after postnatal exposure to benz[a]anthracene and benzo[k]fluoranthene. Environ Toxicol Pharmacol, 2009; 27(2): 253-258. doi: 10.1016/j.etap.2008.11.001
|
| [9] |
IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. IARC Monogr Eval Carcinog Risks Hum, 2010; 92: 1-853.
|
| [10] |
Kang Z, Liu, X B, Yang, C, et al. Effect of ambient PM2.5-bound BbFA and DahA on small airway dysfunction of primary schoolchildren in northeast China. Biomed Res Int, 2019; 2019: 2457964. doi: 10.1155/2019/2457964
|
| [11] |
Malik A I, Rowan-Carroll A, Williams A, et al. Hepatic genotoxicity and toxicogenomic responses in Muta™Mouse males treated with dibenz[a, h]anthracene. Mutagenesis, 2013; 28(5): 543-554. doi: 10.1093/mutage/get031
|
| [12] |
Okona-Mensah K B, Battershill J, Boobis A, et al. An approach to investigating the importance of high potency polycyclic aromatic hydrocarbons (PAHs) in the induction of lung cancer by air pollution. Food Chem Toxicol, 2005; 43(7): 1103-1116. doi: 10.1016/j.fct.2005.03.001
|
| [13] |
Wang X F, Jiang S F, Liu Y, et al. Comprehensive pulmonary metabolome responses to intratracheal instillation of airborne fine particulate matter in rats. Sci Total Environ, 2017; 592: 41-50. doi: 10.1016/j.scitotenv.2017.03.064
|
| [14] |
Xu Y Y, Wang W J, Zhou J, et al. Metabolomics analysis of a mouse model for chronic exposure to ambient PM2.5. Environ Pollut, 2019; 247: 953-963. doi: 10.1016/j.envpol.2019.01.118
|
| [15] |
Zhang Y N, Li Y B, Shi Z X, et al. Metabolic impact induced by total, water soluble and insoluble components of PM acute exposure in mice. Chemosphere, 2018; 207: 337-346. doi: 10.1016/j.chemosphere.2018.05.098
|
| [16] |
He M, Ichinose T, Yoshida S, et al. PM2.5-induced lung inflammation in mice: Differences of inflammatory response in macrophages and type II alveolar cells. J Appl Toxicol, 2017; 37(10): 1203-1218. doi: 10.1002/jat.3482
|
| [17] |
Li R J, Kou X J, Xie L Z, et al. Effects of ambient PM2.5 on pathological injury, inflammation, oxidative stress, metabolic enzyme activity, and expression of c-fos and c-jun in lungs of rats. Environ Sci Pollut Res Int, 2015; 22(24): 20167-20176. doi: 10.1007/s11356-015-5222-z
|
| [18] |
Roberts L D, Souza A L, Gerszten R E, et al. Targeted metabolomics. Curr Protoc Mol Biol, 2012; Chapter 30, Unit 30.2-30.2.24.
|
| [19] |
Sun M, Gao X Q, Zhang D W, et al. Identification of biomarkers for unstable angina by plasma metabolomic profiling. Mol Biosyst, 2013; 9(12): 3059-3067. doi: 10.1039/c3mb70216b
|
| [20] |
Snyder S H, Bredt D S. Biological roles of nitric oxide. Sci Am, 1992; 266(5): 68-71, 74-77. doi: 10.1038/scientificamerican0592-68
|
| [21] |
Virág L, Szabó E, Gergely P, et al. Peroxynitrite-induced cytotoxicity: mechanism and opportunities for intervention. Toxicol Lett, 2003; 11: 113-124, 140-141. doi: 10.1016/S0378-4274(02)00508-8
|
| [22] |
Zhao Y Y, Wang H L, Cheng X L, et al. Metabolomics analysis reveals the association between lipid abnormalities and oxidative stress, inflammation, fibrosis, and Nrf2 dysfunction in aristolochic acid-induced nephropathy. Sci Rep, 2015; 5: 12936. doi: 10.1038/srep12936
|
| [23] |
Walker A K, Jacobs R L, Watts J L, at al. A conserved SREBP-1/ phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell, 2011; 147(4): 840-852. doi: 10.1016/j.cell.2011.09.045
|
| [24] |
Hite R D, Seeds M C, Safta A M, et al. Lysophospholipid generation and phosphatidylglycerol depletion in phospholipase A(2)-mediated surfactant dysfunction. Am J Physiol Lung Cell Mol Physiol, 2005; 288(4): L618-L624. doi: 10.1152/ajplung.00274.2004
|
| [25] |
Hu W Y, Dong T Y, Wang L L, et al. Obesity aggravates toxic effect of BPA on spermatogenesis. Environ Int, 2017; 105: 56-65. doi: 10.1016/j.envint.2017.04.014
|
| [26] |
Sun H, Zhao J, Zhong D, et al. Potential serum biomarkers and metabonomic profiling of serum in ischemic stroke patients using UPLC/Q-TOF MS/MS. PloS one, 2017; 12(12): e0189009. doi: 10.1371/journal.pone.0189009
|
| [27] |
Ebenezer D L, Fu P, Natarajan V. Targeting sphingosine-1-phosphate signaling in lung diseases. Pharmacol Ther, 2016; 168: 143-157. doi: 10.1016/j.pharmthera.2016.09.008
|
| [28] |
Iwatsuki K, Torii K. Peripheral chemosensing system for tastants and nutrients. Curr Opin Endocrinol Diabetes Obes, 2012; 19(1): 19-25. doi: 10.1097/MED.0b013e32834ec7f8
|
| [29] |
Salbaum J M, Kappen C. Genetic and epigenomic footprints of folate. Prog Mol Biol Transl Sci, 2012; 108: 129-158. doi: 10.1016/B978-0-12-398397-8.00006-X
|
| [30] |
Kim J, Hu Z, Cai L, et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature, 2017; 546(7656): 168-172. doi: 10.1038/nature22359
|
| [31] |
Pausch J, Rasenack J, Häussinger D, et al. Hepatic carbamoyl phosphate metabolism. Role of cytosolic and mitochondrial carbamoyl phosphate in de novo pyrimidine synthesis. Eur J Biochem, 1985; 150(1): 189-194. doi: 10.1111/j.1432-1033.1985.tb09006.x
|
| [32] |
Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med, 2009; 30(1-2): 29-41. doi: 10.1016/j.mam.2008.08.003
|
| [33] |
Ni H, Lu L, Deng J, et al. Effects of glutamate and aspartate on serum antioxidative enzyme, sex hormones, and genital inflammation in boars challenged with hydrogen peroxide. Mediators Inflamm, 2016; 2016: 4394695. doi: 10.1155/2016/4394695
|
| [34] |
Waclawiková B, El Aidy S. Role of microbiota and tryptophan metabolites in the remote effect of intestinal inflammation on brain and depression. Pharmaceuticals, 2018; 11(3): 63. doi: 10.3390/ph11030063
|
Figures(5) / Tables(2)
Editorial Office: Frigid Zone Medicine, Editorial Office of Frigid Zone Medicine, Heilongjiang Health Development Research Center, Zhongshan Road 112, Xiangfang District, Harbin, 150036, China
Tel: 0451-87253028
E-mail: editorialoffice@frigidzonemedicine.com
Supported by:
Beijing Renhe Information Technology Co., Ltd.


 Submit
Submit


 DownLoad:
DownLoad: